 IPknot help page
IPknot help page
Contents
Instructions on input
Interpretations of output
References
Instructions on input
You can input either a single RNA sequence (Case 1) or a multiple alignment of RNA sequences (Case 2) into the server.
Case 1: single sequence input
If you want to know a base-paired structure (secondary structure) of a single RNA sequence, enter the sequence in FASTA format into the field as shown below.
Only a sequence like "UUCU..." in 5'–3' direction is also compatible with this interface. Instead, you can submit sequence information by uploading the corresponding FASTA file. Note that the length of the sequence must be less than 1500 nt. If the size of submitted data is over the designated limit, you are recommended to run the stand-alone program instead. Finally, press the "Predict" button, and then you will get a prediction result in the new page.
Case 2: multiple alignment input
To predict a consensus secondary structure of a multiple alignment of RNA sequences, enter the alignment in CLUSTAL W format into the field as follows.
Notice that each block of sequence data in CLUSTAL W must include a line at the bottom that shows the degree of conservation for the columns of the alignment in the block or just an empty line. Also note that each block in CLUSTAL W format must be ended with one or more empty lines. Or alternatively, you can submit an alignment in multiple FASTA format as follows:
The file uploading system is also available. It is to be noted that the length of the alignment, not each sequence, must be less than 1500 nt because our algorithm treats each alignment column as a nucleotide. Then, press the "Predict" button to see a predicted structure in the new page.
Options
There are several parameters that IPknot can adjust.
Level is the number of decompositions of a secondary structure where each decomposed substructure must have no pseudoknots and is said to be at one of the corresponding numbers of decompositions, say, level 1, 2 or 3 in this web tool. Specifically, the decomposition is done in such a way that every base pair at some level must be pseudoknotted with at least one base pair at lower levels. From a different viewpoint, level can be considered as the number of kinds of brackets for indicating base pairs in dot-bracket representation. For example, level 1 uses just one kind of bracket "( )", level 2 uses two kinds of brackets "( )" and "[ ]", and in level 3, three kinds of brackets "( )", "[ ]" and "{ }" are used. Therefore, IPknot of level 1 is an ordinary secondary structure predictor that does not consider pseudoknots like mfold and RNAfold, and it is almost equivalent to CentroidFold. IPknot of level 2 aims to predict nested pseudoknots, whereas IPknot of level 3 seeks to predict pseudoknotted structures with nested pseudoknots. It follows from the way to decompose the original structure that the base pairs at level 3 are less likely to appear than the ones at lower levels.
The server provides three kinds of scoring models that produce base-pairing posterior probabilities. The McCaskill model and the CONTRAfold model take no account of pseudoknotted structures in each decomposed substructure of IPknot, whereas the NUPACK model considers a certain class of pseudoknots. Accordingly, the NUPACK model can be more accurate than the other two models to predict pseudoknotted structures. However, a sequence of length longer than 80 nt is too long for the elaborate NUPACK model to predict fast, and the server rejects the input. Besides, the NUPACK model is not supported for alignment input due to the computational cost.
You can choose whether the base-pairing probabilities of the McCaskill and CONTRAfold models defied over pseudoknot-free structures are refined or not. In the refinement procedure, the base-pairing probabilities are recalculated using the prediction result of the first run of IPknot. It should be noted that the choice of the NUPACK model disallows the refinement due to the computational cost of its iterative use of the NUPACK model.
Level 1 Level 2 Level 3
A weight for each level must be a positive number and is considered similarly. Since the weight represents the rate of true base pairs in each decomposed substructure, it determines prediction accuracy. Note that default values of this web tool are determined by taking several validation results into consideration, which are presented in Supplementary Material of our paper [PubMed]. In general, if the weight increases, the algorithm aims to predict more base pairs and sensitivity of a prediction will get better. On the other hand, if the weight decreases, the algorithm tries to predict less base pairs and positive predictive value will be enhanced. In this sense, the weight is a balanced parameter between sensitivity and positive predictive value. Furthermore, the weight is closely related to the threshold that determines contributable base pairs to improve prediction accuracy. In our framework, the threshold decreases if the weight increases. IPknot adopts the threshold cut technique that discards base pairs with their pairing posterior probabilities less than the threshold, which makes the tool run much faster. Therefore, too large weights are not useful and the server limits the value of each weight to be at most 50.
Interpretations of output
Please see the corresponding case of your input.
Case 1: single sequence input
You find a predicted secondary structure with maximum expected accuracy (MEA). Expected accuracy is the expected number of true predictions measured in base pairs. The MEA structure is first shown in dot-bracket representation as follows.
>MIDV
1 UUCUUUUUUAGUGGCAGUAAGCCUGGGAAUGGGGGCGACCCAGGCGUAUGAACAUAGUGUAACGCUCCCC
1 ....[[..............(((((((]].[[[[[[[.))))))).................]]]]]]].
In the above representation, matching brackets indicate a base pair. Note that different forms of brackets, say "( )" and "[ ]" cross each other, meaning that the structure includes pseudoknots. A similar format to the dot-bracket representation named Vienna is downloadable. In addition, the server can generate BPSEQ format for base-pairing information.
A 2D diagram of the predicted structure along with its arc representation is displayed by running the VARNA program in the background. Note that in the 2D diagram, an A-U pair is indicated by a single line with a bullet, a G-C pair is shown by a double line, and a G-U pair is represented by a single line. In arc representation, the orientation of the RNA sequence is shown by 5' --> 3' at the bottom of the figure.
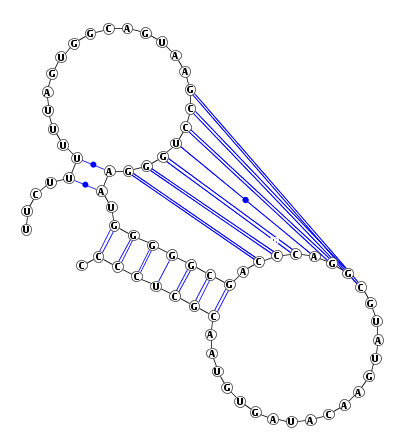
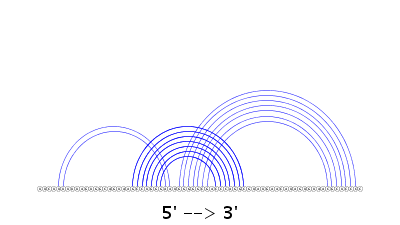
Both the above two drawings are also available as PDF files by pressing the button "PDF."
Case 2: multiple alignment input
The input alignment followed by the MEA common secondary structure is first given as follows (details of MEA are explained in Case 1).
Tomato_mosaic_virus.1 GUGUCUUGGAGCGCGCGGAGUAAACAUAUAUGGUUCAUAUAUGUCCGUAGGCACGUAAAAAAAGCGA
Tobacco_mosaic_virus.1 GUGUCUUGGAUCGCGCGGGUCAAAUGUAUAUGGUUCAUAUACAUCCGCAGGCACGUAAUAAA-GCGA
Rehmannia_mosaic_vir.1 GUGUCUUGGUUCGCGCGGGUCAAGUGUAUAUGGUGCAUAUACAUCCGUAGGCACGUAAUAAA-GCGA
B.pepper.1 GUGUCUUGGAACGCGCGGGUCAAAUAUAAGUGGUUCACUUAUAUCCGUAGGCACGAAAAAUU-GCGU
SS_cons (((((([[[[....((((]]]].(((((((((...))))))))))))))))))).............
Furthermore, a file that contains the predicted consensus structure as well as all input sequences in FASTA format is also downloadable.
The server provides a 2D drawing of the predicted structure and its arc representation using VARNA as shown below.
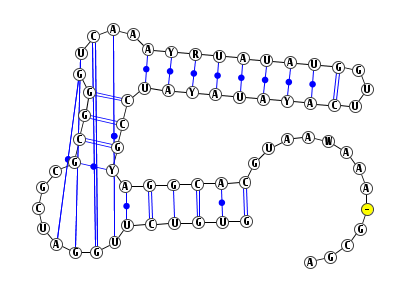
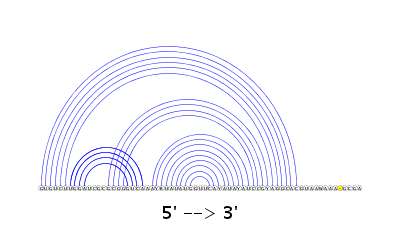
Pressing the button "PDF" generates the corresponding PDF file.
References
If you find IPknot useful, please cite the following article:
- K. Sato, Y. Kato, M. Hamada, T. Akutsu and K. Asai, IPknot: fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming, Bioinformatics, 27, i85-i93, 2011. [PubMed]
IPknot employs RNAfold in the ViennaRNA package (1) with parameters estimated by a Boltzmann likelihood-based method (2), CONTRAfold (3) and NUPACK (4) to compute base-pairing probabilities, and VARNA (5) to visualize predicted structures, whose references are shown below:
- R. Lorenz, S.H. Bernhart, C.H. zu Siederdissen, H. Tafer, C. Flamm, P.F. Stadler and I.L. Hofacker, ViennaRNA Package 2.0, Algorithms Mol. Biol., 6, 26, 2011. [PubMed]
- M. Andronescu, A. Condon, H.H. Hoos, D.H. Mathews and K.P. Murphy, Computational approaches for RNA energy parameter estimation, RNA, 16, 2304-2318, 2010. [PubMed]
- C.B. Do, D.A. Woods and S. Batzoglou, CONTRAfold: RNA secondary structure prediction without physics-based models, Bioinformatics, 22, e90-e98, 2006. [PubMed]
- J.N. Zadeh, C.D. Steenberg, J.S. Bois, B.R. Wolfe, M.B. Pierce, A.R. Khan, R.M. Dirks and N.A. Pierce, NUPACK: analysis and design of nucleic acid systems, J. Comput. Chem., 32, 170-173, 2011. [PubMed]
- K. Darty, A. Denise and Y. Ponty, VARNA: interactive drawing and editing of the RNA secondary structure, Bioinformatics, 25, 1974-1975, 2009. [PubMed]
If you have any questions, please contact Yuki Kato, Kengo Sato.
Graduate School of Medicine, Osaka University, Japan
School of Life Science and Technology, Institute of Science Tokyo, Japan

![[ Powered by Apache ]](../image/apache_pb2.png)