 RactIP help page
RactIP help page
Contents
Instructions on input
Interpretations of output
References
Instructions on input
If you want to know how two RNA sequences interact by base pairing, enter the pair of the sequences in FASTA format into the respective fields as shown below.
Only a sequence like "GGCA..." is also compatible with this interface. Note that each sequence must be put in 5'–3' direction. Instead, you can submit sequence information by uploading two separate FASTA files. You must also note that the sum of the lengths of two sequences must be at most 1000 nt. If the size of submitted data is over the designated limit, you are recommended to run the stand-alone program instead. Finally, press the "Predict" button, and then you can get a prediction result in the new page.
Options
RactIP offers two options.
The server provides two kinds of scoring models that produce internal base-pairing probabilities. Notice that hybridization probabilities related to external base pairs are calculated by a variant of RNAduplex, which is a fixed model in the server.
Internal External
A weight must be a positive number and each weight is considered similarly. Since the weight represents the rate of true internal/external base pairs, it determines the accuracy of joint secondary structure prediction. Note that default values of this web tool are based on the results presented in our paper [PubMed]. In general, if the weight increases, the algorithm aims to predict more base pairs and sensitivity of a prediction will get better. On the other hand, if the weight decreases, the algorithm tries to predict less base pairs and positive predictive value will be enhanced. In this sense, the weight is a balanced parameter between sensitivity and positive predictive value. Furthermore, the weight is closely related to the threshold that determines contributable base pairs to improve prediction accuracy. In our framework, the threshold decreases if the weight increases. RactIP adopts the threshold cut technique that discards base pairs with their pairing posterior probabilities less than the threshold, which makes the tool run much faster. Therefore, too large weights are not useful and the server limits the value of each weight to be at most 50.
Interpretations of output
You find a predicted joint secondary structure with maximum expected accuracy (MEA). Expected accuracy in this case is the expected number of true predictions measured in both internal base pairs and external ones. The MEA structure is first shown in dot-bracket representation as follows.
>R1inv
1 GGCAACGGAUGGUUCGUUGCC
1 ((((((([[[[[[[)))))))
>R2inv
1 GCACCGAACCAUCCGGUGC
1 ((((((]]]]]]]))))))
In the above representation, matching brackets indicate a base pair. Specifically, round brackets "( )" indicate an internal base pair, while square brackets "[ ]" denote an external base pair (binding site). A similar format to the dot-bracket representation named Vienna is downloadable. In addition, the server can generate BPSEQ format for base-pairing information.
The free energy of the predicted joint secondary structure is given by employing RNAeval in the Vienna RNA package.
- Free energy of the predicted joint structure:
Seq. 1: -5.8 kcal/mol; Seq. 2: -5.69 kcal/mol; Hybridization: -3.73 kcal/mol; Joint structure: -15.22 kcal/mol
A drawing of the predicted joint structure in arc representation is displayed by running the VARNA program in the background. Note that blue arcs represent internal base pairs, whereas red arcs stand for external interactions. The orientation of each RNA sequence is shown by 5' --> 3' at the bottom of the figure.
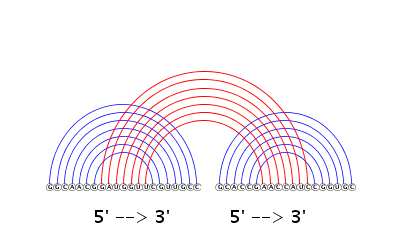
The above drawing of original size is also available in PDF format by pressing the button "PDF."
References
If you find RactIP useful, please cite the following article:
- Y. Kato, T. Mori K. Sato, S. Maegawa, H. Hosokawa and T. Akutsu, An accessibility-incorporated method for accurate prediction of RNA-RNA interactions from sequence data, Bioinformatics, 33, 202–209, 2017. [PubMed]
- Y. Kato, K. Sato, M. Hamada, Y. Watanabe, K. Asai and T. Akutsu, RactIP: fast and accurate prediction of RNA-RNA interaction using integer programming, Bioinformatics, 26, i460-i466, 2010. [PubMed]
RactIP employs CONTRAfold (1), and RNAfold and RNAduplex in the ViennaRNA package (2) with parameters estimated by a Boltzmann likelihood-based method (3) to compute base-pairing probabilities, and VARNA (4) to visualize predicted joint structures, whose references are shown below:
- C.B. Do, D.A. Woods and S. Batzoglou, CONTRAfold: RNA secondary structure prediction without physics-based models, Bioinformatics, 22, e90-e98, 2006. [PubMed]
- R. Lorenz, S.H. Bernhart, C.H. zu Siederdissen, H. Tafer, C. Flamm, P.F. Stadler and I.L. Hofacker, ViennaRNA Package 2.0, Algorithms Mol. Biol., 6, 26, 2011. [PubMed]
- M. Andronescu, A. Condon, H.H. Hoos, D.H. Mathews and K.P. Murphy, Computational approaches for RNA energy parameter estimation, RNA, 16, 2304-2318, 2010. [PubMed]
- K. Darty, A. Denise and Y. Ponty, VARNA: interactive drawing and editing of the RNA secondary structure, Bioinformatics, 25, 1974-1975, 2009. [PubMed]
If you have any questions, please contact Yuki Kato, Kengo Sato.
Graduate School of Medicine, The University of Osaka, Japan
School of Life Science and Technology, Institute of Science Tokyo, Japan

![[ Powered by Apache ]](../image/apache_pb2.png)